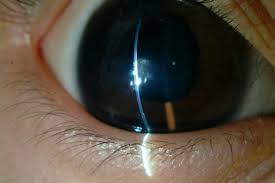
Peters anomaly is a congenital corneal disease caused by abnormal embryogenesis of the anterior segment of the eyeball. The pathology is caused by mutations in FOXC1, PAX6, PITX2 and CYP1B1, which occur sporadically or are inherited from parents. Symptoms of the disease: white opacity above the pupil and iris, microcornea and microphthalmos, Anophthalmos decreased visual acuity of varying severity. Diagnosis is made based on the results of biomicroscopy, OCT and ultrasound of the eye, tonometry, autorefractometry. Treatment is performed in the ophthalmic surgery department using penetrating keratoplasty, which is supplemented with antiglaucoma operations if indicated.
General information
The disease is baptized afterwards the German ophthalmologist Alfred Peters who first labeled it in 1906. There are currently 35,000 people with this disease in the world, and about 700 patients in Russia. The incidence of Peters’ anomaly is 1.5-3 cases per 100,000 people. No gender or racial differences in incidence have been found. Without treatment, the disease ends with a sharp decrease or complete loss of vision, so it remains relevant in modern practical ophthalmology .
Reasons
Peters anomaly is formed during the intrauterine period of a child’s development. It looks through alterations of homeobox protein sequence (FOXC1, PAX6, PITX2), which regulate cellular differentiation and growth of multicellular organisms. These courses are necessary used for the formation of various organs of the body including the eyeball. Rarely, Peters disease occurs with a mutation of the CYP1B1 gene, which encodes cytochrome P450 1B1 and is responsible for the embryonic development of the anterior part of the eye and dry eye syndrom .
No risk factors have been identified that contribute to the occurrence of genetic mutations. Rarely, Peters anomaly is hereditary. Damaged homeobox genes are transmitted to the child from parents in an autosomal dominant manner. CYP1B1 pathologies are inherited in an autosomal recessive manner.
Pathogenesis
Normally, embryogenesis of the eyeball begins at the end of the 2nd week of gestation, when paired optic pits appear in the ectoderm. The sclera, cornea iris & vitreous body are placed unhappy in the 4th to 6th week of emergent development and consist of mesenchyme. During this period all layers of the cornea are formed:
epithelium, Bowman of membrane, stroma, Descemet of membrane and endothelium.
In Peters anomaly, the formation of the anterior structures of the eye is disrupted. In all cases of the disease, there is clouding of the central cornea – the section responsible for the refraction of light rays and their conduction to the lens. Clouding is accompanied by pathologies of the inner corneal layers – Descemet’s membrane and endothelial cells, as well as adhesions between the corneoscleral and iris membranes of the eye.
Classification Several approaches to classifying the disease have been described in medical literature. One of the first was the 1974 classification proposed by W. Townsend et al., who divided Peters’ anomaly into 3 types: Type 1 – isolated corneal opacity ( leukoma ), Type 2 – leukoma with adhesions between the cornea and lens, Type 3 – opacity with Rieger’s mesodermal dysgenesis. In modern ophthalmology, another classification is used, which includes 2 options:
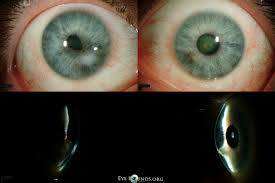
- Mesodermal (type 1) is a classic “soft” form of Peters difference. It is manifested by structural abnormalities in the central parts of the cornea, connective tissue adhesions between the corneal membrane and the iris.
- Ectodermal (type 2) is a more severe form of the disease, which combines the signs of the previous variant and other eye lesions: clouding and displacement of the lens, underdevelopment of the anterior chamber, iridocorneolenticular adhesions.
Symptoms of Peters anomaly
Signs of the disease are present in the child from birth. The main clinical manifestation is a visible white opacity in the center of the cornea, which covers the pupil and iris. In type I Peters anomaly, the defect looks like a translucent cloud and is surrounded by strands of the iris that cross the anterior chamber of the eye. It also feel like cataract. In type II disease, a dense white opacity is observed, which spreads over the entire surface of the cornea.
Many children have concomitant eye development disorders. Often, a decrease in the diameter of the cornea (microcornea), flattening of the corneal membrane, and small eye sizes ( microphthalmos ) are determined like myopia . Less mutual stand chorioretinal coloboma a fragmented trendy the eye sheaths, aniridia a wide-ranging absence of the iris and sclerocornea a prolix clouding of the cornea, in which it is externally indistinguishable from the sclera.
In 50% of patients with Peters anomaly, there is a significant deterioration in vision , in 25% – blindness . In 60-80% of cases, the pathology is bilateral, which aggravates the severity of symptoms. As the child grows older, additional symptoms appear: inability to follow a toy and parents’ faces with the eyes, spontaneous squinting and twitching of the eyes, strabismus, a habit of bending the head low during games and looking at pictures.
Complications
One of the most common consequences of the disease is glaucoma , which is observed in 30-70% of cases. Its occurrence is caused by mesenchymal dysgenesis – abnormalities in the development of tissues in the area of the anterior chamber angle, which result in disturbances in the outflow of aqueous humor. Increased intraocular pressure is fraught with optic neuropathy and optic nerve atrophy , which causes vision loss and eye strain .
In 35% of cases, Peters anomaly is accompanied by malformations of the central nervous system, cardiovascular and genitourinary systems. It is possible to develop Peters-plus syndrome, in which eye damage is supplemented by cleft palate , color blindness , facial dysmorphism, conjunctivitis, hearing impairment, congenital shortening of the limbs. Rarely, children with such a diagnosis are diagnosed with psychomotor development disorders .
Diagnostics
Congenital corneal opacity in an infant requires consultation with a pediatric ophthalmologist . In the presence of concomitant developmental defects, the help of a clinical geneticist will be required. During the initial examination, attention is paid to the size of the eyeballs, the synchronicity of their movements, the presence and size of a white spot on the cornea. To establish a diagnosis, special ophthalmological diagnostic methods will be required, such as:
- Ultrasound biomicroscopy . The study shows a dense and thickened cornea, the presence of iridocorneal contact along its entire length, and a hyperechoic structure of the iris. In type 2 anomalies, multiple adhesions and fibrous processes are determined.
- Optical coherence tomography . The technique is used for detailed examination of the eye, including when the transparency of optical media is reduced. OCT allows measuring the thickness of the cornea determining its shape and layered structure and assessing the features of vascularization.
- Ultrasound of the eye . Ophthalmoechography provides detailed information about the condition of the structures of the posterior segment of the eye, which cannot be seen with ophthalmoscopy due to the opacity of the cornea. Ultrasound B-scan shows the eye muscles and other structures of the orbit.
- Tonometry of the eye . The study is conducted to measure intraocular pressure and timely diagnosis of glaucoma. In children a non contact tonometry method is used which provides a quick and painless assessment of IOP values.
- Visual acuity assessment . In small children, standard visometry with tables is impossible, so a special method is used – skiascopy . Computer autorefractometry is performed to assess the refractive power of the eye structures .
Differential diagnostics
Peters anomaly must be distinguished from other forms of congenital (primary) corneal opacity: Fuchs endothelial dystrophy , posterior polymorphic dystrophy, isolated peripheral sclerocornea. Differential diagnostics are also carried out with TORCH infections (rubella, toxoplasmosis, cytomegalovirus), perinatal eyeball trauma, cystinosis and mucopolysaccharidoses .
Treatment of Peters anomaly
Surgery is the only effective method for correcting eyeball malformations. Treatment time is determined individually taking into account the severity of the anomaly the presence of concomitant disorders and the visual acuity index. Since Peters anomaly causes irreversible damage to the cornea, penetrating keratoplasty is used to eliminate the defect . During the operation, the affected area of the eye shell is replaced with a donor transplant.
Glaucoma is a contraindication to reconstructive surgery. Such patients first undergo sinus trabeculectomy to create a new pathway for the outflow of ocular fluid and reduce IOP. Penetrating keratoplasty is performed on average 3 months after compensation of intraocular pressure. According to indications, local ophthalmic hypotensive drugs are prescribed in the postoperative period .
Prognosis and prevention
The success rate of keratoplasty is 30-60%. In 22-67% of cases, repeated surgeries are required due to graft opacity. The best results are achieved when surgery is performed in the first six months of the baby’s life. Most patients achieve visual acuity greater than 0.3, which improves the child’s adaptation and intellectual development. Prevention of Peters’ anomaly has not been developed due to the spontaneous nature of gene mutations and the rarity of the disease.